Distrofias musculares en la infancia: Distrofinopatías
CONCEPTO
Causadas por ausen
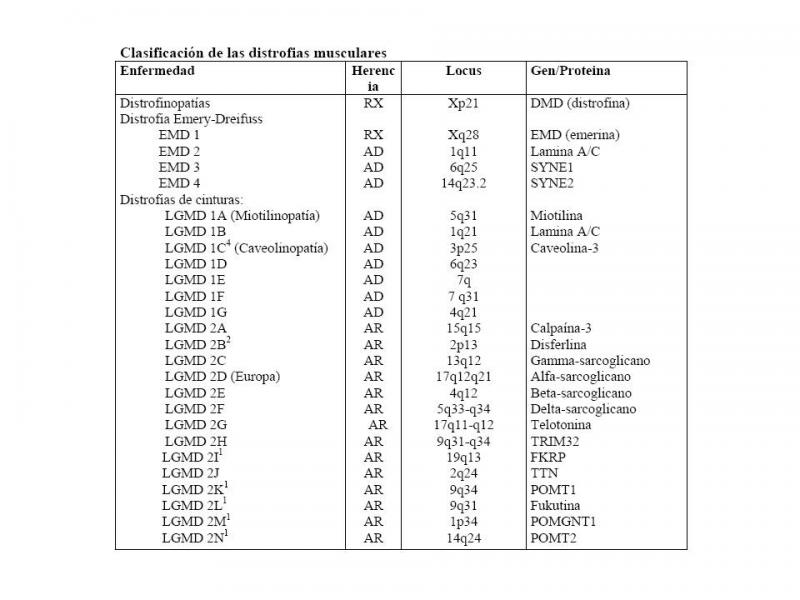
cia (Duchenne), disminución o expresión anómala (Becker) de distrofina, proteína estructural que une el citoesqueleto y matriz extracelular, lo que protege a la célula muscular durante la contracción de membrana y que se encuentra fundamentalmente en músculo esquelético; también en músculo liso, corazón y sistema nervioso central(SNC). La distrofina está codificada por un gen de gran tamaño DMD situado en locus Xp21(Cohn et al;2010). (Ver “Clasificación de las distrofias musculares.jpg”).
Se reconocen dos cuadros clínicos: enfermedad de Duchenne y enfermedad de Becker. Otros fenotipos intermedios caracterizados por hiperCKemia con leve o nula afectación de músculo esquelético: miocardiopatía dilatada ligada al X sin afectación muscular, miopatía del cuádriceps y síndrome de mialgias-calambres(Dubrovsky et al;2007).
ENFERMEDAD DE DUCHENNE
Aspectos generales
Herencia recesiva ligada al cromosoma X(33% mutaciones de novo). Es la distrofia más frecuente de inicio en la infancia. Incidencia:1:3.500 varones.
Portadoras de enfermedad: El 70% de las mujeres portadoras presentan hiperCKemia leve-moderada. Generalmente son asintomáticas, aunque en un 10% pueden presentar debilidad leve proximal y anomalías en el electromiograma (EMG), cardiacas o cognitivas que no suelen afectar a la calidad de vida(Colomer et al;2011).
Clínica
- Debilidad muscular rápidamente progresiva de predominio proximal. Se pueden distinguir 3 fases:
- Fase de inicio: Los síntomas aparecen antes de los 5 años (generalmente 2-4), pero debido a la maduración del SNC el paciente sigue desarrollando funciones musculares. Puede haber retraso en la adquisición de ítems madurativos tanto motores como del lenguaje. Al iniciar la marcha, ésta es torpe, con caídas frecuentes y dificultad para correr o subir escaleras. Aparecen contracturas y la debilidad es fundamentalmente a nivel de la musculatura de la cintura pélvica y flexora cervical; mientras que la afectación de la cintura escapular es menos manifiesta y la musculatura facial está prácticamente conservada. Por la debilidad adoptan en bipedestación una actitud en hiperlordosis, con tendencia a marcha de puntillas y contracturas aquíleas secundarias (por debilidad de musculatura extensora de cadera y rodilla).
- Fase de meseta (“plateau”): Estancamiento en adquisición de habilidades motoras; entre los 6 y los 8 años.
- Fase de empeoramiento: Afectación progresiva de musculatura distal, miembros superiores y pares craneales (disfagia). La base de sustentación va aumentando, la hiperlordosis se acentúa y desarrollan pie varo. La debilidad progresa hasta perder la deambulación antes de los 12 años, lo que provoca un empeoramiento de las deformidades esqueléticas (escoliosis y contracturas) y de la función respiratoria (patología restrictiva). Asocian problemas nutricionales (sobrepeso o desnutrición); fracturas por disminución de densidad ósea y problemas de relación social.
- Afectación no musculo-esquelética: Alteración cognitiva; suelen presentar valores de cociente intelectual más bajos de lo normal (CI:75-90), trastornos del aprendizaje y alteraciones conductuales. Miocardiopatía dilatada, frecuente aunque los síntomas sean tardíos. Afectación del sistema digestivo: estreñimiento, reflujo gastro-esofágico.
Exploración física
En las primerias fases son frecuentes las hipertrofias musculares especialmente en pantorrillas: pseudo-hipertrofia de gemelos (dureza elástica a la palpación, secundaria a infiltración por tejido graso y fibrótico). ROT disminuidos, van desapareciendo progresivamente; el aquíleo es el último en desaparecer. Signo de Gowers (dificultad al ponerse de pie, que el niño realiza apoyando los brazos en sus rodillas y luego en muslos hasta alcanzar la posición erecta:“autoescalamiento”). Marcha miopática (“ansarina”) con actitud hiperlordótica. Con la evolución aparecen contracturas y atrofia de la musculatura proximal.
Pronóstico
Morbilidad: Secundaria a complicaciones respiratorias (75%) o cardiacas (25%). Mortalidad: Clásicamente la esperanza de vida era menor de 20 años, aunque el tratamiento integral ha hecho que la supervivencia media alcance la tercera década de la vida.
ENFERMEDAD DE BECKER
Incidencia:1:30.000 varones. Aunque el fenotipo es más heterogéneo, en general se diferencia por el inicio más tardío (a partir de 5 años) y la evolución más lenta (marcha independiente más allá de los 16 años). La esperanza de vida supera los 40 años; mortalidad secundaria a trastorno cardíaco(Donoso et;2008).
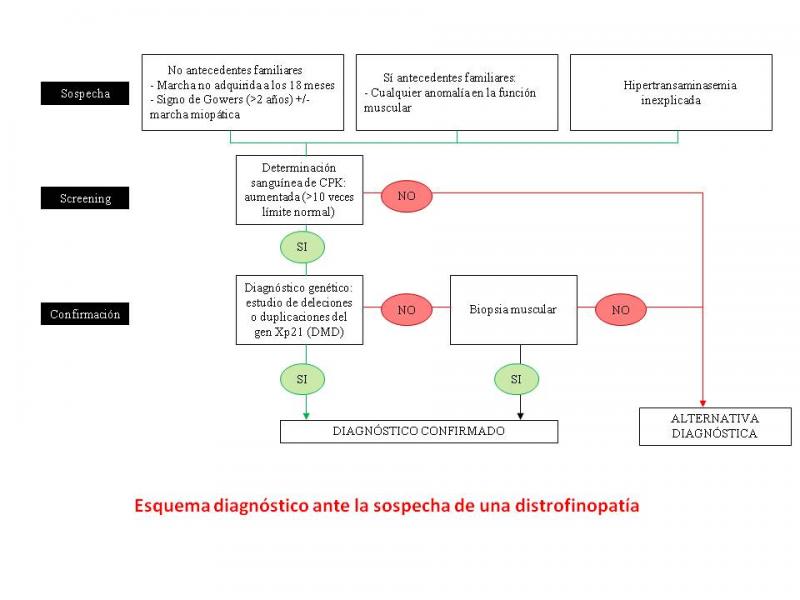
DIAGNÓSTICO
Recomendaciones generales
Ante la sospecha, se recomienda inicialmente el diagnóstico mediante el estudio de las mutaciones del gen; sólo en caso de no encontrarlas se recomienda la biopsia muscular. En aquellos pacientes diagnosticados anteriormente mediante biopsia se recomienda el estudio del gen para consejo genético y posibles tratamientos génicos.
Enzimas musculares
HiperCKemia con niveles de 10 a 50 veces superior a lo normal. El aumento de otras enzimas séricas (aldolasa, LDH, transaminasas) puede inducir a realización de biopsias hepáticas por un diagnóstico erróneo de hepatitis crónica.
Electromiograma
Puede ser útil en el diagnóstico diferencial. Trazado mixto con aumento de la actividad de inserción y fibrilaciones, las ondas positivas son menos frecuentes. Los potenciales de unidad motriz son pequeños y de corta duración, aunque hasta un 30% son largos y con potenciales tardíos.
Biopsia muscular
Las alteraciones varían según progresa la enfermedad. En estadios iniciales hay variación en el tamaño de las fibras y áreas focales de degeneración y regeneración. Según progresa la enfermedad la regeneración va siendo menor, aparecen signos de necrosis y el músculo es sustituido por tejido conectivo y adiposo. No es obligada, aunque está indicada si el estudio genético es negativo; y puede ser útil en el diagnóstico diferencial. El estudio inmunohistoquímico y mediante Western-Blot de la distrofina es el único método que permite diferenciar la forma Duchenne de la Becker (aparte de la evolución clínica): ausencia completa de distrofina en Duchenne, distrofina anómala en Becker.
Estudios genéticos
Es la técnica gold-standard para el diagnóstico, detección de portadoras y diagnóstico prenatal. Estudio de las anomalías del gen de la distrofina: 65-70% se debe a deleciones de uno o más exones, el 5-10% a duplicaciones y el resto se debe a mutaciones puntuales (inserciones, mutaciones “non-sense” o “stop codón”). Estas mutaciones puntuales en ocasiones no se detectan con las técnicas moleculares habituales (MLPA y PCR múltiple) y es necesario una secuenciación del gen completo. Es posible el diagnóstico prenatal(Erazo-Torricelli et al;2004)( López-Hernández et al;2010).
TRATAMIENTO
No existe tratamiento curativo aunque, un manejo multidisciplinar mejora la calidad de vida y altera el curso natural de la enfermedad aumentando la esperanza de vida (Strober et al;2006):
TRATAMIENTO ACTUAL:
Corticoides
Son la única medicación que ha demostrado ralentizar el curso de la enfermedad: mejoran la fuerza, función muscular, reducen el riesgo de escoliosis, mejoran la función pulmonar y cardíaca y parecen prolongar la deambulación hasta 3 años. No hay un consenso claro sobre el régimen a emplear, aunque en las últimas revisiones recomiendan comenzar en la fase de “plateau”, con prednisona dosis única 0,75 mg/kg/día (máximo:30-40 mg/día) o deflazacort 0,9 mg/kg/día (máximo:40 mg/día). Aparentemente es igual de efectiva prednisona administrada únicamente en fines de semana (10 mg/kg/semana)(Escolar et al; 2011). El tratamiento suele mantenerse hasta la pérdida de la deambulación. Es necesaria una vigilancia estrecha de posibles efectos secundarios como retraso crecimiento, cambios conductuales, HTA, hiperglucemia, síntomas gastrointestinales, fracturas óseas o síndrome Cushing. Si excesivos efectos adversos valorar cambiar tipo de corticoide, reducir 33% dosis o emplear esquemas intermitentes (10-20). Puede ser útil asociar un antisecretor ácido y vitamina D. Últimos estudios apuntan que la asociación con bifosfonatos puede aumentar la esperanza de vida en aquellos con corticoterapia crónica(Gordon et al;2011).
Tratamiento integral
Incluye: Ejercicio aeróbico submáximo, tratamiento rehabilitador y traumatológico de contracturas y escoliosis, control cardiológico (ecocardiografía y electrocardiograma cada 1-2 años), asistencia respiratoria mediante fisioterapia respiratoria, sistemas de ventilación nocturna (BIPAP) y técnicas de asistencia a la tos, así como controles de la función pulmonar anualmente. Otros aspectos importantes que se deben manejar: Psicosocial, gastrointestinal, nutricional (evitar sobrepeso y desnutrición), manejo de disfagia y dolor. Contraindicados determinados procedimientos anestésicos: relajantes despolarizantes del músculo (succinilcolina) y no aconsejados los anestésicos inhalados (riesgo de rabdomiolisis)(Bushby et al;2010).
PERSPECTIVAS DE TRATAMIENTO
Hay grandes avances en los últimos años en terapias basadas en la modificación de la expresión de distrofina mediante la alteración del ARNm que han ofrecido esperanzadores resultados: terapia de omisión exónica (“exón-skipping”) mediante el uso de oligonucleótidos antisentido u omisión de codones de paro (“stop-codón”). Otros tratamientos en investigación: reemplazo del gen (terapia génica con vectores virales, plásmidos o células madre), administración de proteínas compensadoras (utrofina) o estabilizadores de membrana (poloxamer)(López-Hernández et al;2009)(Nelson et al;2009).
BIBLIOGRAFÍA
- Bushby K. (2010). Diagnosis and management of Duchenne muscular dystrophy. Lancet Neurol. 9:77-93
- Cohn RD, Campbell KP. (2000). Molecular basis of muscular dystrophies. Muscle Nerve. 23:1456-71.
- Colomer J, Nascimiento A. (2011). Patología neuromuscular. En: Campistol J (Ed), Neurología para pediatras: Enfoque y manejo práctico, (pp369-388). Madrid: Editorial Medica Panamericana
- Donoso MA, Garzón M, Martínez B. (2008). Distrofias musculares. Miotonía congénita. En: Verdú A, García A, Martínez B (Ed), Manual de Neurología Infantil, (pp 696-703). Madrid:Publimed.
- Dubrovsky AL, Tarauto AL. (2007). Miopatías. En: Fejerman N, Álvarez E (Ed), Neurología Pediátrica, (pp544-576). Argentina: Editorial Médica Panamericana
- Erazo-Torricelli R. (2004). Actualización en distrofias musculares. Rev Neurol. 39:860-871.
- Escolar DM, Hache LP, Clemens PR, Cnaan A, McDonald CM, Viswanathan V et al. (2011). Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy.Neurology. 77:444-52.
- Gordon KE, Dooley JM, Sheppard KM, MacSween J, Esser MJ. (2011). Impact of bisphosphonates on survival for patients with Duchenne muscular dystrophy. Pediatrics. 127:353-358.
- López-Hernández LB. (2009). Distrofia muscular de Duchenne: actualidad y perspectivas de tratamiento. Rev Neurol. 49:369-375.
- López-Hernández LB, Ayala-Madrigal ML, van Heudsen D, Estrada-Mena FJ, Canto P, Sandóval-Ramírez L et al. (2010). Mejoras en el diagnóstico de distrofinopatías: ¿qué hemos aprendido después de 20 años? Rev Neurol. 52:239-249.
- Nelson SF. (2009). Emerging genetic therapies to treat Duchenne muscular dystrophy. Curr Opin Neurol. 22:532-538
- Strober JB. (2006). Therapeutics in Duchenne muscular dystrophy. NeuroRx. 3:225-34.

{kind=link}
{kind=link}