Déficit de biotinidasa
Los errores innatos del metabolismo son enfermedades monogénicas de herencia autosómica recesiva en su mayoría (Wolf, 2007). Dentro de ellos tenemos la deficiencia o ausencia de biotinidasa, conocida también anteriormente como deficiencia múltiple de carboxilasa de comienzo tardío. La causa es un defecto en la absorción o el transporte de la biotina, que normalmente se produce por una alteración de la biotinidasa o de la holocarboxilasa sintetasa.
La biotinidasa es una glicoproteína monomérica con un peso molecular de 76 a 77 kDa (Hart 1991) y tiene 9 isoformas; posee una actividad enzimática elevada en suero, hígado, riñón y glándula suprarrenal y además también la encontramos en el jugo pancreático, en la orina humana y en las células secretoras (Wolf 1984, Heard 1985, De Felice 1995). Tiene diferentes funciones como son la actividad hidrolítica, biotiniltransferasa, lipoaminidasa y como proteína transportadora de biotina (Hymes 1997).
- Metabolismo de la biotina
La biotina pertenece al grupo de las vitaminas hidrosolubles del complejo B y está involucrada en importantes procesos metabólicos debido a su papel como grupo prostético de las siguientes enzimas:
a)acetil CoA carboxilasa: implicada en la biosíntesis de los ácidos grasos. (Cardellá 1999)
b)piruvato carboxilasa: interviene en la gluconeogénesis y en el ciclo del ácido cítrico.
c)propionil CoA carboxilasa: interviene en el ciclo de los ácidos tricarboxílicos. (Stryer 1982)
d)metil crotonil CoA carboxilasa: interviene en el metabolismo de la leucina.
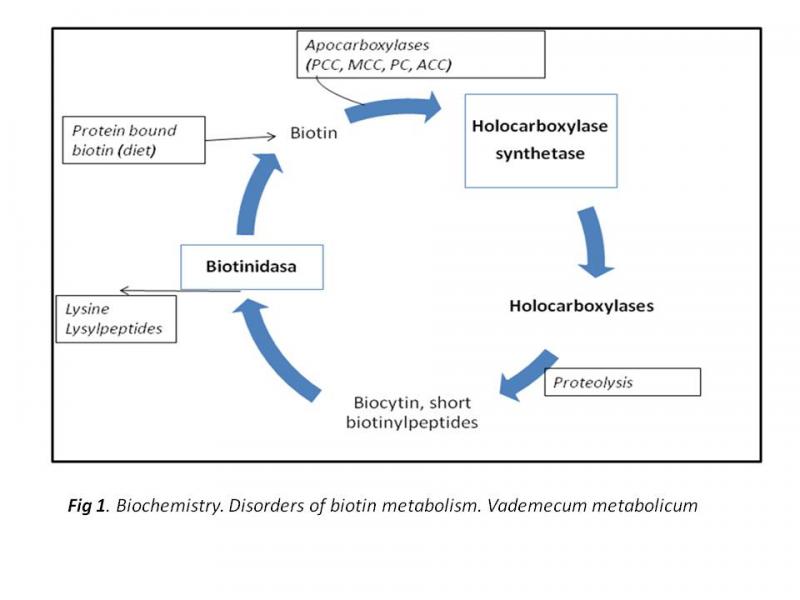
La biotina se une al sitio activo de estas enzimas y funciona como acarreador de CO2. Las carboxilasas se sintetizan como apocarboxilasas, carentes de biotina y la forma activa se produce por la unión covalente de la biotina al grupo amino de un residuo de lisina de la apocarboxilasa, reacción catalizada por la holocarboxilasa sintetasa. El paso final de la degradación de las carboxilasas es la rotura de la fracción biotinil del grupo amino de la lisina que es catalizada por la biotinidasa y acaba con la liberación de la biotina libre, la cual puede ser nuevamente reciclada. La biotina regula a nivel postranscripcional la expresión de la propionil-CoA carboxilasa y a nivel transcripcional, la expresión de la holocarboxilasa sintetasa. Además de este papel, la biotina está involucrada en otras áreas del metabolismo.
La deficiencia de biotina se ha reportado en pacientes sometidos a una alimentación parenteral total, en personas que ingieren grandes cantidades de clara de huevo crudo, en niños con desnutrición energético-proteíca severa y en personas con errores innatos del metabolismo. (Rodríguez 2000)
- Epidemiología y cuadro clínico
La incidencia de la enfermedad a nivel mundial se estima que es 1:110000 para la deficiencia total y 1:60000 para la deficiencia parcial. (Wolf 1983)(Bhardwaj 2010).
La deficiencia de biotina se caracteriza por una encefalopatía metabólica de inicio agudo, retraso psicomotor, convulsiones refractarias a tratamiento antiepiléptico, hipotonía, alopecia, rash cutáneo y alteraciones del sistema inmunológico en aquellos pacientes con una deficiencia grave de biotina sin tratamiento administrado. Una vez los problemas de visión, la pérdida de cabello y el retraso madurativo están presentes, son irreversibles aunque iniciemos el tratamiento con la biotina.
Las características más tardías, propias de los déficits parciales, son hipotonía, ataxia, retraso madurativo, problemas de visión, pérdida de audición y anomalías cutáneas, seguido por debilidad, paresia espástica y descenso en la agudeza visual. En estos pacientes todos estos síntomas se agudizan sobretodo en los momentos de estrés.
La deficiencia de biotina se debe sospechar cuando se presentan los síntomas siguientes y se confirma a través de los tests enzimáticos. (Wolf 2010).
Los síntomas neurológicos más comunes son las convulsiones y la hipotonía (Wolf et al 1983, 1985, 1988 y 1995). Las convulsiones suelen ser mioclónicas, pero también pueden ser tónico-clónicas generalizadas y focales; algunos niños tienen espasmos infantiles (Salbert et al).
La mayoría de los niños con déficit de biotinidasa muestran anomalías en el sistema nervioso central en la RM cerebral o la TC. (Wolf et al 1983, Wastell et al 1988, Salbert et al 1993, Lott et al 1993, Grunewald et al 2004), como por ejemplo: edema cerebral, señal atenuada de la sustancia blanca, atrofia cerebral y aumento del tamaño de los ventrículos. Estos hallazgos pueden mejorar o normalizarse después del tratamiento con biotina.
Bioquímicamente la enfermedad se caracteriza por acidosis metabólica, con hiperamoniemia y aumento del lactato, así como aciduria orgánica (aumento de alanina y disminución de la carnitina), aunque la ausencia de estos hallazgos no excluye el diagnóstico.
- Diagnóstico
Los individuos con una deficiencia severa de biotinidasa tienen menos de un 10% de la media normal de la actividad enzimática en suero. Aquellos que tienen un déficit parcial de biotinidasa tienen una actividad enzimática entre un 10-30%. Ambas alteraciones, tanto la severa como la parcial, se detectan en el screening metabólico neonatal. En el screening metabólico se utiliza una pequeña cantidad de sangre obtenida de la punción del talón del bebé para realizar un test colorimétrico que mide la actividad de la biotinidasa (Heard 1984, Wolf 1985, Heard 1986, Wolf 1991).
Si en el screening metabólico los resultados están alterados se procede a la obtención de una muestra de plasma/suero del bebé afecto.
Asimismo se puede detectar la actividad enzimática de la biotinidasa en muestras de leucocitos y fibroblastos. (Wolf and Secor Mc Voy 1983).
También está disponible un test de genética molecular de BTD, único gen cuyas mutaciones se sabe que se relacionan con el déficit de biotinidasa. Este test es de utilidad cuando el resultado de los análisis es ambiguo. El diagnóstico prenatal para embarazos de riesgo precisa primero de la identificación de las mutaciones causantes de la enfermedad.
- Evolución de la enfermedad
Las personas que son diagnosticadas antes de desarrollar los síntomas y que son tratadas con biotina tienen un desarrollo normal. Los problemas neurológicos ocurren sólo en aquellos pacientes que tienen síntomas recurrentes y alteraciones a nivel metabólico antes de iniciar el tratamiento.
En el déficit severo de biotinidasa los síntomas en aquellos pacientes no tratados normalmente aparecen entre la semana de vida y los 10 años, con una media de edad de 3 meses y medio (Wolf et al 1985). Aproximadamente el 76% de los niños no tratados con un déficit severo de biotinidasa tienen una pérdida de audición neurosensorial, que normalmente no se resuelve una vez iniciado el tratamiento, pero que permanece estable cuando éste se inicia. (Wolf 2002)
Algunos niños con déficit de biotinidasa severo permanecen asintomáticos hasta la adolescencia, momento en el que desarrollan una pérdida repetina de visión con neuropatía óptica progresiva y paraparesia espástica (Ramaekers et al 1992, 1993, Lott 1993). Tras unos meses de tratamiento con biotina, las alteraciones visuales se resuelven y la paraparesia espástica mejora. Hay, por otro lado, pacientes en los que se detecta alteración en el screening metabólico al nacimiento pero que permanecen asintomáticos a lo largo de toda su vida (Wolf 1997, Baykal 2005). Los pacientes con déficit parcial de biotinidasa, pueden manifestar sólo síntomas en los momentos de estrés o de infecciones.
- Tratamiento
El resultado de un estudio en niños con déficit de biotinidasa indica que el tratamiento con biotina es efectivo para prevenir los síntomas (Möslinger 2001, Weber 2004). Todos los niños con déficit severo de biotinidasa mejoran cuando inician tratamiento con biotina 5-10 mg al día por vía oral. Todos los pacientes diagnosticados deben recibir tratamiento con biotina durante toda la vida.
Dosis recomendadas:
- Déficit biotinidasa: 5-10 mg/día de biotina
Algunos de los síntomas como la atrofia óptica, la pérdida de audición o el retraso psicomotor pueden no ser reversibles, por lo que deben ser controlados por especialistas para poder paliar los déficits, con intervenciones oftalmológicas, audífonos o implantes cocleares y pautas para disminuir el efecto del retraso psicomotor, en caso de que sean necesarias.
Las alteraciones bioquímicas y las convulsiones desaparecen rápidamente tras iniciar el tratamiento con biotina, seguidas por la mejoría de las alteraciones cutáneas. En los niños con alopecia, tras un periodo de semanas a meses vuelve a evidenciarse un crecimiento del pelo. La atrofia óptica y la pérdida de audición pueden ser resistentes al tratamiento, sobre todo si hay un largo periodo de tiempo entre el inicio de los síntomas y el inicio del tratamiento. Una vez iniciado el tratamiento estos síntomas permanecen estables.
No se precisa de ninguna restricción dietética, únicamente se debe evitar el huevo crudo, puesto que contiene avidina, proteína que se une a la biotina y disminuye la biodisponibilidad de la vitamina.
- Controles evolutivos
En todos los pacientes con déficit de biotinidasa se deben hacer controles anuales por oftalmología, otorrinolaringología y neurología.
- Diagnóstico diferencial
Déficit de biotina: puede ser diagnosticado al realizar una historia alimentaria, por ejemplo pacientes con nutrición parenteral sin suplemento de biotina.
Déficit aislado de carboxilasas: el análisis de ácidos orgánicos en orina es útil para diferenciar el déficit aislado de carboxilasa del déficit múltiple de carboxilasas que ocurre en la deficiencia de biotinidasa y en el déficit de holocarboxilasa sintetasa:
- El metabolito más elevado en orina en el déficit de biotinidasa, en el déficit de holocarboxilasa sintetasa, en el déficit aislado de beta-metilcrotonil-CoA carboxilasa y en la deficiencia adquirida de biotina es el beta-hidroxisovalerato.
- Por otro lado, la presencia de concentraciones elevadas de lactato, metilcitrato y beta-hidroxipropionato en orina son indicativos del déficit múltiple de carboxilasas.
El deficit multiple de carboxilasas responde a tratamiento con biotina, por el contrario el déficit aislado de carboxilasas, no responde a este tratamiento. Para llegar al diagnóstico de déficit aislado de carboxilasa tenemos que demostrar la disminución de la actividad enzimática bien en los leucocitos de sangre periférica (para las carboxilasas mitocondriales) o en fibroblastos.
Déficit de holocarboxilasa sintetasa: responde al tratamiento con biotina. Los síntomas son muy similares a los del déficit de biotinidasa, aunque la sintomatología puede iniciarse de forma más temprana, antes de los 3 meses.
Al diagnóstico llegamos mediante el análisis específico de la actividad de cada enzima.
Bibliografía
Baykal T, Gokcay G, Gokdemir Y, Demir F, Seckin Y, Demirkol M, Jensen K, Wolf B. Asymptomatic adults and older siblings with biotinidase deficiency ascertained by family studies of index cases. J Inherit Metab Dis. 2005;28:903–12.
Bhardwaj P, Kaushal RK, Chandel A. Biotinidase deficiency: A treatable cause of infantile seizures. J Pediatr Neurosci 2010;5:82-3
Grunewald S, Champion MP, Leonard JV, Schaper J, Morris AA. Biotinidase deficiency: a treatable leukoencephalopathy.Neuropediatrics. 2004;35:211–6
Hart PS, Hymes J ,Wolf B. Isoforms of human serum biotinidase. Clin Chim Acta 1991; 197 : 257-64.
Heard GS, Secor McVoy JR, Wolf B. A screening method for biotinidase deficiency in newborns. Clin Chem. 1984;30:125–7.
Heard GS, Wolf B, Jefferson LG, Weissbecker KA, Nance WE, McVoy JR, Napolitano A, Mitchell PL, Lambert FW, Linyear AS. Neonatal screening for biotinidase deficiency: results of a 1-year pilot study. J Pediatr. 1986;108:40–6.
Lott IT, Lottenberg S, Nyhan WL, Buchsbaum MJ. Cerebral metabolic change after treatment in biotinidase deficiency. J Inherit Metab Dis. 1993;16:399–407
Möslinger D, Stockler-Ipsiroglu S, Scheibenreiter S, Tiefenthaler M, Muhl A, Seidl R, Strobl W, Plecko B, Suormala T, Baumgartner ER. Clinical and neuropsychological outcome in 33 patients with biotinidase deficiency ascertained by nationwide newborn screening and family studies in Austria. Eur J Pediatr. 2001;160:277–82.
Weber P, Scholl S, Baumgartner ER. Outcome in patients with profound biotinidase deficiency: relevance of newborn screening. Dev Med Child Neurol. 2004;46:481–4.
Wolf B, Heard GS, McVoy JS, Raetz HM. Biotinidase deficiency: The possible role of biotinidase in the processing of dietary protein-bound biotin. J Inher Metab Dis 1984; 7: 121-2.
Wolf B, Heard GS, Jefferson LG, Proud VK, Nance WE, Weissbecker KA. Clinical findings in four children with biotinidase deficiency detected through a statewide neonatal screening program. N Engl J Med. 1985b;313:16–9
Wolf B. Worldwide survey of neonatal screening for biotinidase deficiency. J Inherit Metab Dis. 1991;14:923–7
Cardellá RL, Hernández FR. Metabolismo de la glucosa. En: Cheping N, editor. Bioquímica Médica. C. Habana: Ciencias Médicas. 1era ed, 1999. Vol. III. p. 830-47.
Heard GS, Grier RE, Weiner DL, McVoy JR, Wolf B. Biotinidase deficiency. Ann NY Acad Sci 1985; 447:252-62.
De Felice C, Hayakawa K, Tanaka T, Terentieva E. Biotinidase activity in the urine of healthy subjects. Nephron 1995; 70:115.
Hymes J, Fleischhaver K, Wolf B. Biotinidase in serum and tissues. Methods Enzymol 1997; 279: 422-34.
Rodríguez Meléndez Rocío. Importancia del metabolism de la biotina. Rev Invest Clin 2000; 52 (2): 194-199.
Wastell H, Dale G, Bartlett K. A sensitive fluorimetirc rate for biotinidase using a new derivative of biotin, biotinyl-6-aminoquinoline. Anal Biochem. 1984;140:69–73.
Wolf B: Biotinidase deficiency. In GeneClinics: Medical Genetics Knowledge Base (online database). Seattle, University of Washington, October 22, 2007. Available at: http: //www.geneclinic.org
Wolf B, Grier RE, Parker WD, Goodman SI, Allen RJ. Deficient Biotinidase activity in late -onset multiple carboxylase deficiency. N Engl J Med 1983; 308: 161.
Wolf B, Secor McVoy J. A sensitive radioassay for biotinidase activity: deficient activity in tissues of serum biotinidase-deficient individuals. Clin Chim Acta. 1983;135:275–81
Wolf B, Heard GS, Weissbecker KA, McVoy JR, Grier RE, Leshner RT. Biotinidase deficiency: initial clinical features and rapid diagnosis. Ann Neurol. 1985c;18:614–7.
Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 2010;100:6–13
Wolf B, Spencer R, Gleason T. Hearing loss is a common feature of symptomatic children with profound biotinidase deficiency. J Pediatr. 2002b;140:242–6.
Stryer L. Glycolysis. En: Stryer L, editor. Biochemistry. Madrid-Barcelona: Reverte, S.A. 2 da ed. 1982. p. 307-27.
Salbert BA, Pellock JM, Wolf B. Characterization of seizures associated with biotinidase deficiency. Neurology.1993b;43:1351–5
Ramaekers VT, Brab M, Rau G, Heimann G. Recovery from neurological deficits following biotin treatment in a biotinidase Km variant. Neuropediatrics. 1993;24:98–102.
Ramaekers VT, Suormala TM, Brab M, Duran R, Heimann G, Baumgartner ER. A biotinidase Km variant causing late onset bilateral optic neuropathy. Arch Dis Child. 1992;67:115–9

{kind=link}
{kind=link}