Trastornos miotónicos en la infancia
CONCEPTO DE MIOTONÍA
Dificultad en relajación muscular tras una contracción voluntaria. En la exploración se puede provocar mediante percusión con martillo en eminencia tenar, lengua o muslo. Suele mejorar tras movimiento (fenómeno de “calentamiento”), y empeorar tras reposo. Este fenómeno se observa al solicitar al paciente que cierre fuertemente el puño durante unos segundos y posteriormente lo abra de manera repetida; encontrando mejoría progresiva con la repetición. Se diferencia de la paramiotonía, donde no se observa este fenómeno de “calentamiento”, empeorando tras el ejercicio. Son varios los trastornos que pueden manifestarse con miotonía en la infancia.
Además la presencia en electromiograma(EMG) de descargas miotónicas características (salva espontánea de alta frecuencia con disminución gradual) es clave para el diagnóstico.
DISTROFIAS MIOTÓNICAS
DISTROFIA MIOTÓNICA TIPO 1 (ENFERMEDAD STEINERT)
Aspectos Generales
Enfermedad multisistémica de herencia autosómica dominante (AD) con expresividad variable debida a la expansión inestable del triplete CTG en gen DMPK. Un individuo normal tiene ≤34 repeticiones; 35-49 repeticiones supone un riesgo de transmisión; son necesarias >50 repeticiones para presentar síntomas. Existe correlación positiva entre el número de repeticiones con la intensidad y aparición precoz de los síntomas; con lo que la presentación clínica es un continuum de gravedad relacionado con el número de tripletes (Colomer et al;2011).
- Incidencia:1/8.000.
- Fenómeno de anticipación: Aumento de repetición de tripletes en cada generación; lo que provoca sintomatología cada vez más grave y precoz.
Forma infantil
1. Clínica
750-1200 repeticiones. Debutan generalmente con trastornos del aprendizaje secundarios a déficit cognitivos o trastornos de coordinación; porque las manifestaciones musculares suelen ser leves al inicio. Posteriormente van desarrollando el cuadro clásico caracterizado por:
- Debilidad de musculatura distal y facial (cara larga y delgada por debilidad de maseteros y temporales, junto con labio inferior evertido: aspecto de “atónito”). La atrofia suele aparecer a partir de la tercera década.
- Miotonía, no suele aparecer hasta la edad adulta.
- Afectación cardiaca(>50%). Alteraciones de conducción: bloqueo aurículo-ventricular y/o bloqueo de rama. Hasta un 10% presentan muerte súbita. También pueden asociar anomalías estructurales.
- Trastornos de sueño(80%): Alteración primaria de la regulación del sueño que provoca excesiva somnolencia diurna, mayor porcentaje de sueño REM y movimientos periódicos de piernas(Yu et al;2011).
- Signos extra-musculares: Afectación cognitiva estable no evolutiva y déficits atencionales. Cataratas subcapsulares posteriores precoces. Alteraciones de la motilidad gastrointestinal (estreñimiento, disfagia, litiasis biliar…). Alteraciones conductuales (impulsividad). Trastornos endocrinos (atrofia gonadal, resistencia a insulina, anomalía suprarrenal). Alopecia fronto-parietal en hombres adultos. Neuropatía axonal periférica. Hipogammaglobulinemia.
2. Pronóstico
Mortalidad aumentada secundaria a neumonías aspirativas, dificultad respiratoria, arritmias (muerte súbita): esperanza de vida de 48-55 años.
3. Diagnóstico
- Sospechado ante síntomas característicos en familiares: miotonía, debilidad muscular o cataratas.
- HiperCKemia moderada.
- EMG: descarga miotónica característica, indicado en caso de duda.
- Diagnóstico mediante cuantificación del triplete CTG en el gen DMPK.
4. Tratamiento
Sintomático según las distintas manifestaciones: Para el fenómeno miotónico estudios recientes han mostrado eficacia de clomipramina y mexiletina (Logigian et al;2010). Importante el seguimiento cardiológico así como endocrinológico. Riesgo de efectos secundarios graves con algunos agentes anestésicos.
Forma congénita
1. Clínica
>1200 repeticiones. La transmisión es casi siempre por vía materna.
- Prenatal: Síntomas de hipocinesia fetal (polihidramnios, menor movilidad fetal, deformidades de los pies). Debido al fenómeno miotónico en útero materno son frecuentes los problemas perinatales: prematuridad (50%), atonía uterina, abortos, partos distócicos.
- Periodo neonatal: Hipotonía, debilidad (llamativa diplejía facial, labio superior con forma de “V” invertida, paladar ojival), dificultad respiratoria (ventilación mecánica) y dificultades para la succión y deglución. Suelen asociar deformidades esqueléticas (artrogriposis, pie zambo) y hernia diafragmática.
- Periodo postnatal. Los niños que sobreviven al periodo neonatal presentarán hipotonía grave durante los primeros meses, retraso motor (la mayoría consiguen la marcha autónoma con más de 2 años), retraso mental significativo. Posteriormente se añaden las características de la forma clásica de manera más intensa: Debilidad (marcha hasta los 20-25 años), fenómeno miotónico aparece a partir de los 10 años, trastornos cardiacos, cataratas…(Dubrovsky et al;2007)
2. Diagnóstico
Sospechado por clínica y presencia de la enfermedad en la madre. Confirmación definitiva por estudio de genética molecular. EMG en periodo neonatal no suele ser útil.
3. Tratamiento
Tratamiento de soporte en periodo neonatal. Posteriormente, tratamiento sintomático al igual que en la forma infantil.
4. Pronóstico
El fallecimiento en etapa neonatal alcanza el 40% por afectación respiratoria. Una vez superado el periodo neonatal la supervivencia es similar a la forma infantil.
DISTROFIA MIOTÓNICA TIPO 2 (MIOPATÍA MIOTÓNICA PROXIMAL)
Enfermedad multisistémica, AD, debida a la expansión del tetra-nucleótido CCTG en el gen ZNF9. La sintomatología es similar a la DM tipo1, aunque se diferencia en que la intensidad es más leve, se afecta fundamentalmente la musculatura proximal y suele aparecer a partir de la tercera década de la vida (aunque la miotonía y mialgias pueden aparecer en la primera década)(Donoso et al;2008).
El diagnóstico se realiza mediante estudio de genética molecular. EMG en caso de duda. Otras datos evocadores: HiperCKemia, Hipogammaglobulinemia.
El tratamiento es sintomático. En caso de dolor, puede ser útil el empleo de mexiletina o carbamacepina.
MIOTONÍAS NO DISTRÓFICAS
MIOTONÍA CONGÉNITA
Producida por mutaciones en el gen del canal del cloro (CLCN1). La herencia puede ser AD (forma Thomsen) o AR (forma Becker).
1. Clínica
Espectro fenotípico variable desde miotonía leve sin repercusión funcional a miotonía grave, incapacitante; asociada a debilidad muscular crónica y mialgias. Comienza en la infancia temprana. Las formas dominantes suelen ser más leves y tempranas. Los pacientes presentan hipertrofia muscular generalizada (apariencia “hercúlea”)(Lossin et al;2008)(Holding-Jorgensen et al;2005).
2. Diagnóstico
CK sérica normal (o levemente aumentada). EMG: descargas miotónicas características. Genética molecular para el estudio de mutaciones del gen.
3. Tratamiento
El empleo de fármacos antimiotónicos (mexiletina, imipramina y taurina) pueden estar indicados en casos de afectación funcional o mialgias asociadas a miotonía.
PARAMIOTONÍA CONGÉNITA (MIOTONÍA PAROXÍSTICA)
AD. Episodios de rigidez y debilidad desencadenados por frío o por ejercicio, que aparecen en la primera década de la vida(Aparicio et al;1994).
MIOTONÍA AGRAVADA POR POTASIO (K)
AD. Episodios de rigidez fluctuantes que se desencadenan por ingesta de K.
SÍNDROME SCHWARTZ-JAMPEL (MIOTONÍA CONDRODISTRÓFICA)
AR. Miotonía grave que asocia talla baja, deformidades óseas, contracturas articulares, anomalías faciales (rigidez de la musculatura de la cara) y anomalías oculares.
BIBLIOGRAFÍA
- Aparicio JM. (1994). Distrofias musculares, miopatías congénitas y síndromes miotónicos. Medicine. 6:2239-50.
- Colomer J, Nascimiento A. (2011). Patología neuromuscular. En: Campistol J (Ed), Neurología para pediatras. Enfoque y manejo práctico, (pp369-388). Madrid: Editorial Medica Panamericana.
- Donoso MA, Garzón M, Martínez B. (2008). Distrofias musculares. Miotonía congénita. En: Verdú A, García A, Martínez B (Ed), Manual de Neurología Infantil, (pp 696-703). Madrid:Publimed.
- Dubrovsky AL, Tarauto AL. (2007). Miopatías. En: Fejerman N, Álvarez E (Ed), Neurología Pediátrica, (pp544-576). Argentina: Editorial Médica Panamericana.
- Holding-Jorgensen E. (2005). Phenotypic variability in myotonia congenita. Muscle Nerve. 32:19-34.
- Logigian EL, Martens WB, Moxley RT, McDermott MP, Dilek N, Wiegner AW et al. (2010). Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology. 74:165-170.
- Lossin C, George AL Jr. (2008). Myotonia Congenita. Adv Genet. 63:25-55.
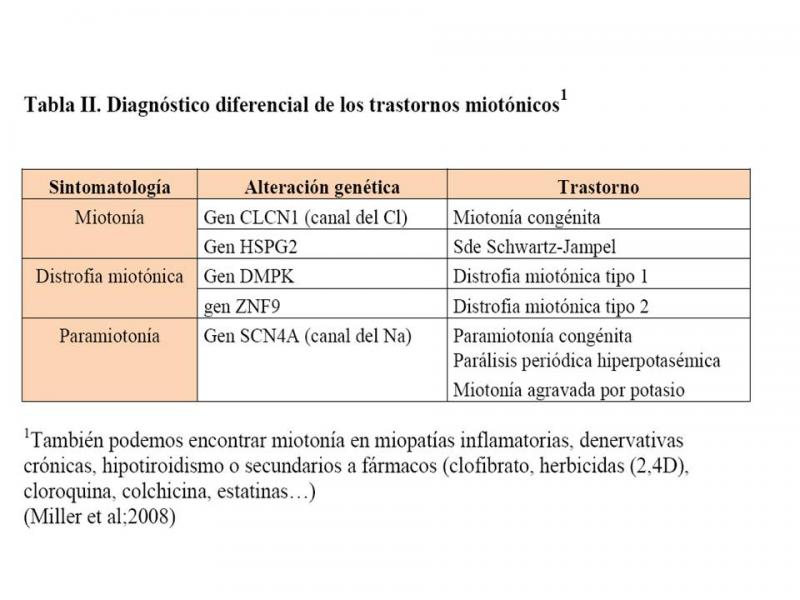
- Miller TM. (2008). Differential diagnosis of myotonic disorders. Muscle Nerve.37:293-9
- Yu H, Laberge L, Jaussent I, Bayard S, Scholtz S, Morales R et al. (2011). Daytime Sleepiness and REM Sleep Characteristics in Myotonic Dystrophy: A Case-Control Study. Sleep. 34:165-170.

{kind=link}