Generalidades de los Trastornos metabólicos neonatales con afectación neurológica
INTRODUCCIÓN
Los errores congénitos del metabolismo (ECM) son un grupo de enfermedades muy heterogéneo, que pueden manifestarse con una gran variedad de síntomas neurológicos. Su incidencia real se desconoce realmente (aproximadamente 1:1000 recién nacidos vivos), si bien gracias al cribado neonatal se conoce la de determinadas entidades como la fenilcetonuria (1:12.000), hipotiroidismo (1:2.000) e incluso de algunas acidurias orgánicas (Campistol et al; 2007).
Su diagnóstico es difícil debido a que son muchos los ECM que pueden manifestarse en periodo neonatal (al menos el 50%) y que el recién nacido (RN) tiene un repertorio limitado de respuestas a la enfermedad grave, por lo que los ECM en esta etapa no se manifiestan con una sintomatología específica y con frecuencia sus síntomas se confunden con los de otros procesos más frecuentes (por ejemplo: sepsis).
La rentabilidad diagnóstica de las pruebas complementarias en muchas ocasiones es baja, pero el diagnosticar una ECM desde el periodo neonatal es crucial para poder ofrecer un pronóstico adecuado, consejo genético, evitar a los padres una “odisea diagnóstica” y en algunos casos ofrecer tratamientos metabólicos específicos curativos.
A pesar de que los trastornos metabólicos son una causa no infrecuente de sintomatología neurológica en periodo neonatal; no hay que olvidar que la primera actuación ante un recién nacido con síntomas neurológicos es la de descartar alteraciones más comunes y potencialmente graves como sepsis, encefalopatía hipóxico-isquémica, hemorragia del sistema nervioso central y patología cardiopulmonar (Sanjurjo et al; 2003).
CRIBADO NEONATAL AMPLIADO
El uso de técnicas de espectrometría de masas en tándem (MS/MS) ha permitido ampliar el número de enfermedades detectables mediante cribado neonatal. Esto es claramente útil para detectar enfermedades de expresión neonatal lo que permite un diagnóstico y tratamiento precoz; al igual que para detectar enfermedades de expresión en periodos más tardíos (como infancia o adolescencia) que permita un adecuado seguimiento, prevención y tratamiento precoz de descompensaciones.
En el estudio más amplio realizado (250.000 recién nacidos alemanes) con cribado neonatal de 23 ECM (aminoacidopatías, acidurias orgánicas y defectos de beta oxidación de los ácidos grasos), la sensibilidad fue mayor del 95% y la tasa de valor predictivo positivo alcanzó el 11%. Se identificaron 106 recién nacidos con ECM; de los cuales 61 se beneficiaron de un tratamiento específico por lo que permanecieron asintomáticos sin secuelas (Schulze et al; 2003). Otros estudios también han demostrado que el cribado neonatal ampliado es una técnica eficaz y costo-efectiva en la identificación precoz de ECM, lo que permite iniciar precozmente el tratamiento y reducir la morbi-mortalidad (McCabe et al; 2008).
Los programas de cribado neonatal, iniciados en nuestro país en 1968 se han ido implantando, aunque con grandes diferencias entre las distintas comunidades autónomas. Actualmente en muchas de estas comunidades se han iniciado programas de cribado neonatal ampliado. Por ejemplo, en la Comunidad de Madrid el cribado neonatal incluía 5 enfermedades (hipotiroidismo congénito, fenilcetonuria, hiperplasia suprarrenal congénita, fibrosis quística y hemoglobinopatías), pero desde Octubre de 2011 se ha ampliado hasta 17 incluyendo aminoacidopatías, acidurias orgánicas y defectos de beta oxidación.
SOSPECHA DE ECM
- Sospecha de enfermedad metabólica por los antecedentes
- Antecedentes familiares.
• Dado que la mayoría de los ECM son autosómicos recesivos, puede sospecharse si los padres son consanguíneos o pertenecen a una comunidad étnica o geográfica cerrada.
• Antecedente de un hermano muerto por una enfermedad aguda no definida (“sepsis”) o por síndrome muerte súbita.
• Antecedente de otros familiares con trastornos no explicados: enfermedad neurológica progresiva, abortos repetidos, infertilidad…
- Enfermedades maternas durante el embarazo.
• Síndrome de HELLP (hemólisis, elevación de las enzimas hepáticas, plaquetopenia) puede estar relacionado con deficiencia de ácidos grasos de cadena larga (LCHAD) del feto. En estos casos es el estrés metabólico provocado por el ECM del feto lo que precipita la clínica en la madre. En los neonatos con madres que han sufrido esta patología habría que valorar la determinación de acilcarnitinas.
• Embriopatía por fenilcetonuria materna no tratada se relaciona con abortos de repetición o hijos con clínica de retraso mental, microcefalia congénita, defectos cardiacos, bajo peso (CIR) y dismorfia facial.
• Retraso del crecimiento intrauterino como forma inicial de manifestación de enfermedades mitocondriales o defectos de la síntesis de colesterol.
- Sospecha de enfermedad metabólica por la clínica
Ya hemos comentado que debemos tener un alto índice de sospecha para poder diagnosticar metabolopatías en el periodo neonatal porque el sistema nervioso inmaduro del RN reacciona de forma inespecífica ante cualquier estrés. La presentación de enfermedades metabólicas no es específica y puede simular cuadros de sepsis, o hemorragia cerebral. Sin embargo, son muy sugestivas las siguientes presentaciones:
- Deterioro neurológico progresivo y rápido en ausencia de antecedentes perinatales patológicos.
- El deterioro inesperado y “misterioso” de un niño tras un periodo inicial normal (sugiere un ECM de tipo intoxicación).
- Dificultad para controlar con tratamiento convencional ciertas situaciones patológicas.
- Afectación multiorgánica especialmente afectación hepática, cardiaca y encefalopatía; aunque también se puede afectar el sistema renal, coagulación, ocular…
- Alteración del ritmo respiratorio (hiperpneas, apneas) en ausencia de trastornos cardiopulmonares.
- A veces el síntoma inicial es una infección; pero, que una vez resuelta, no resuelve el cuadro de afectación general; como ocurre en la galactosemia clásica que se acompaña con frecuencia de infección por Escherichia Colli.
CLASIFICACIÓN
Clínicamente es difícil distinguir unas enfermedades metabólicas de otras ya que todas comparten signos y síntomas comunes. Además constantemente se describen en la literatura médica nuevos ECM así como nuevas formas de presentación de trastornos metabólicos conocidos. Hay varias clasificaciones que pueden ayudarnos a distinguirlos:
1. - Clasificación por grupos básicos
- Intoxicación:
Se caracterizan por un acúmulo de un metabolito tóxico próximos al lugar de bloqueo metabólico, por lo que tras un periodo libre de síntomas se producen los síntomas.
Son recién nacidos aparentemente sanos en los que, tras un intervalo libre de pocas horas-días, inician una clínica de deterioro neurológico progresivo. Al principio los síntomas son más inespecíficos: vómitos, irritabilidad, somnolencia, alteraciones vegetativas y especialmente hipotonía.
Posteriormente se instaura un grave deterioro neurológico progresivo (convulsiones, alteración de la conciencia y coma), con una situación de “catástrofe metabólica” (acidosis metabólica, cetosis, hipoglucemia, ácido láctico aumentado, hiperamoniemia, trombopenia, coagulopatía). En resumen, se caracterizan por un deterioro inesperado y “misterioso” de un niño tras un periodo inicial normal.
Muchas de estas enfermedades son tratables siempre que el tratamiento se instaure precozmente para evitar una acumulación del metabolito tóxico.
En este grupo se incluyen los defectos del metabolismo intermediario:
• Aminoacidopatías
• Acidurias orgánicas
• Trastornos del ciclo de la urea
• Galactosemia.
• Alteraciones del metabolismo de los neurotransmisores
• Otros: intoxicación por metales.
- Alteración del metabolismo energético
Se caracterizan por un error en el metabolismo intermediario que origina un déficit en la producción o en la utilización de la energía necesaria para un funcionamiento adecuado. Esto se traduce en un defecto en múltiples órganos y sistemas, especialmente hígado, músculo esquelético, corazón y cerebro, siendo raros la letargia y el coma. No hay un periodo libre de síntomas y la intensidad es más variable por lo que la presentación clínica es menos evocadora.
Los síntomas son heterogéneos e incluyen: estancamiento ponderal, alteración del ritmo respiratorio, convulsiones, hipotonía, miopatía, cardiopatía, fallo renal, arritmias, defectos de conducción, insuficiencia cardiaca, muerte súbita, malformaciones cerebrales y dismorfias. En resumen, se caracterizan por un recién nacido con fallo multiorgánico.
En este gruo se incluyen:
• Trastornos mitocondriales: trastornos del metabolismo del piruvato, trastornos de la cadena respiratoria mitocondrial y trastornos del ciclo de Krebs.
• Defectos de la oxidación de los ácidos grasos.
• Glucogenosis, defecto de la gluconeogénesis, hiperinsulinismo congénito.
• Trastornos del metabolismo de la creatina.
- Alteración de moléculas complejas
Se caracterizan por una alteración en la síntesis o catabolismo de las moléculas complejas.
Los síntomas son más permanentes o lentamente progresivos sin relación con eventos intercurrentes ni con la dieta. Las manifestaciones son más variadas e inespecíficas (cataratas, ascitis, ictericia, edema, hepatoesplenomegalia, trastorno de la coagulación, dismorfia) lo que dificulta su diagnóstico.
En este grupo se incluyen:
• Enfermedades lisosomales.
• Peroxisomales.
• Errores innatos del metabolismo del colesterol.
• Síndrome de glicoproteínas deficientes en carbohidratos (CDG)
• Trastorno del tráfico intracelular (deficiencia de -1-antitripsina).
2,- Clasificación de Saudubray
Saudubray combina claves clínicas y analíticas para clasificar las enfermedades metabólicas neonatales y con ello facilitar su estudio (Saudubray et al; 2002)(Fernandes et al; 2006).
- Grupo I: Deterioro neurológico tipo intoxicación sin deshidratación ni cetoacidosis grave:
• Aminoacidopatías tipo leucinosis (o enfermedad de la orina con olor a jarabe de arce).
- Grupo II. Cetoacidosis e hiperamoniemia:
• Deterioro neurológico tipo intoxicación: acidurias orgánicas.
• Deterioro neurológico tipo déficit de energía (con disfunción hepática o síntomas cardiacos): defectos de oxidación de los ácidos grasos o defectos de cetogénesis.
- Grupo III: Acidosis láctica con distrés neurológico tipo déficit energético:
• Acidosis lácticas congénitas: alteraciones del metabolismo del piruvato, de la cadena respiratoria mitocondrial o del ciclo de Krebs.
• Defecto múltiple de carboxilasas.
- Grupo IV: Deterioro neurológico sin cetoacidosis:
A) Grupo IV-a: Tipo intoxicación con hiperamoniemia:
• Trastornos del ciclo de la urea.
• Defectos de la oxidación de los ácidos grasos.
B) Grupo IV-b: Sin hiperamoniemia, con hipotonía grave, crisis, mioclonías:
• Hiperglicinemia no cetósica.
• CDG.
• Déficit de sulfito oxidasa.
• Peroxisomales.
• Trastornos de la cadena respiratoria mitocondrial.
• Trastornos de los neurotransmisores.
• Trastornos de la síntesis del colesterol.
• Epilepsia dependiente de piridoxina o de piridoxal-fosfato.
- Grupo V: Con afectación hepática
A) Grupo V-a: Hipoglucemia recurrente con hepatomegalia
• Glucogenosis tipo 1 y 3
• Deficiencia de fructosa difosfatasa (trastorno de la gluconeogénesis)
• Trastornos de oxidación de los ácidos grasos
• Hiperinsulinismo
B) Grupo V-b: Insuficiencia hepática
• Galactosemia
• Tirosinemia tipo 1 (tras 3 semanas)
• Hemocromatosis neonatal
• Trastorno de la cadena respiratoria mitocondrial
• Deficiencia de transaldolasa (vía pentosa-fosfato)
C) Grupo V-c: Ictericia colestásica (generalmente asociada a fallo de medro e hipotonía)
• Deficiencia de 1-antitripsina
• Enfermedad de Byler (colestasis intrahepática familiar)
• ECM de los ácidos biliares
• Peroxisomal
• Niemann-Pick tipo C
• CDG
• Trastornos del metabolismo del colesterol
• LCHAD.
D) Grupo V-d: Signos sugerentes de enfermedad de depósito (facies tosca, visceromegalias, ascitis, hidrops fetal, macroglosia, cambios en huesos, mancha roja cereza, linfocitos vacuolados):
• Enfermedades lisosomales.
• CDG.
3.- Clasificación por la clínica
- Síntomas neurológicos
• Deterioro neurológico (coma, letargia). Ver “Metabolopatía neonatal con deterioro neurológico”
• Convulsiones. Ver “Metabolopatía neonatal con convulsiones”.
• Hipotonía. Ver “Metabolopatía neonatal con hipotonía”.
- Dismorfia Ver “Metabolopatía neonatal con dismorfia”.
- Síntomas hepáticos: Ver “Metabolopatía neonatal con alteración hepática”
- Síntomas cardiacos: Ver “Metabolopatía neonatal con alteración cardíaca”
CLAVES DIAGNÓSTICAS DE ECM
Aunque en muchas ocasiones la expresión de los diferentes ECM son similares hay algunas claves clínicas y analíticas que nos permiten diferenciarlos (Franco-Fernández et al;2008)(Lyon et al;2006):
1.- Claves clínicas (Nyhan et al; 2010)
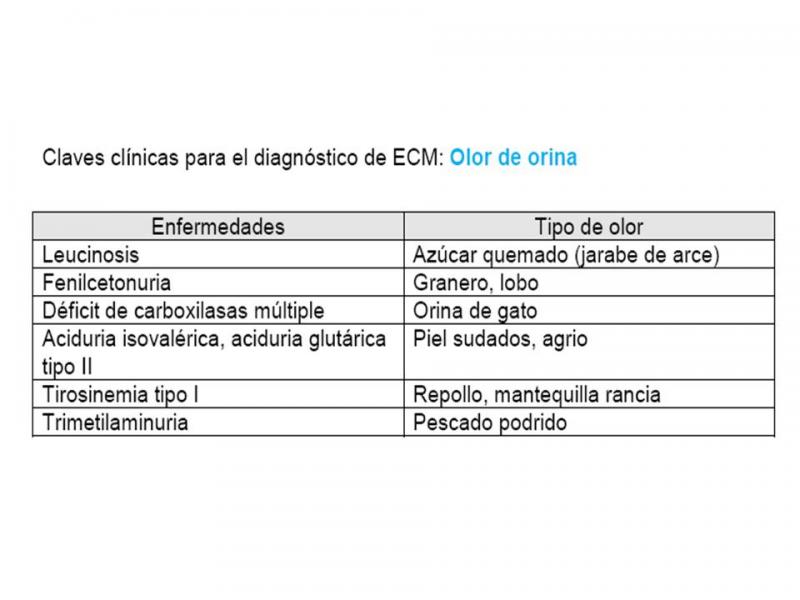
- Olor
Olor anormal corporal o de orina (en muestra fresca) por acumulación de metabolitos volátiles.
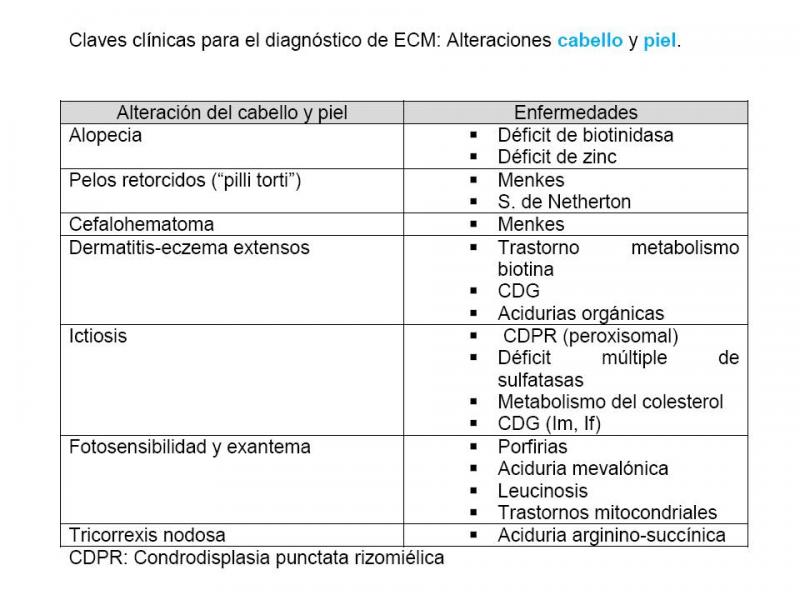
- Alteraciones del cabello y piel
- Otras claves clínicas
- ECM que presentan lesiones prenatalmente:
• S. de Zellweger y otras peroxisomales
• Trastorno del metabolismo del piruvato.
• Aciduria glutárica tipo 2
• Aciduria 3-OHisobutírica
• Deficiencia de sulfito oxidasa
• Hiperglicinemia no cetósica
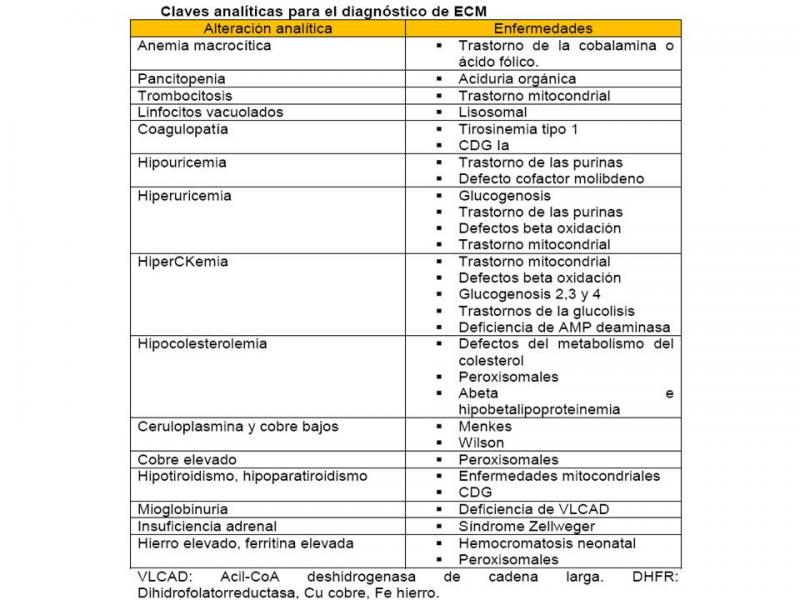
2.- Claves analíticas
La determinación de hemograma, ionograma, bioquímica, lactato, amonio y gasometría sanguínea es esencial en la evaluación inicial de un paciente con sospecha de ECM (Ver “Diagnóstico de metabolopatía en el recién nacido”).
Algunas claves analíticas evocadoras son (González et al;2010) (Garcia-Cazorla et al;2008):
2.1.- Acidosis metabólica con anion gap aumentado (>16): sugestivo de acidurias orgánicas. Muchos recién nacidos con vómitos se diagnostican erróneamente de estenosis pilórica; cualquier neonato con vómitos y acidosis con anión GAP aumentado debe hacernos sospechar un trastorno metabólico.
2.2.- Acidosis láctica: son muchas las enfermedades no metabólicas, como la hipoxemia y la hipoperfusión producidas por sepsis, enfermedad cardiaca, pulmonar o convulsiones, que producen acidosis láctica. Elevaciones moderadas (3-6 mmol/l) se observan frecuentemente en los ECM (como hipermoniemias y acidemias orgánicas); en cambios niveles mayores (>10 mmol/l) son frecuente en situaciones de hipoxia. Unas cifras de pH normal no excluyen una hiperlactacidemia puesto que éste no se modificará mientras las concentraciones de ácido láctico no lleguen a 5 mmol/l.
o Acidosis láctica congénita: (especialmente si aumento de lactato en LCR y sangre): alteraciones del metabolismo del piruvato y trastornos de la cadena respiratoria mitocondrial.
o Acidosis láctica secundaria: defectos de la oxidación de los ácidos grasos (DBO), acidurias orgánicas, trastornos del ciclo de la urea, glucogenosis (I, III) y trastornos de gluconeogénesis. Muchos ECM producen acidosis en estadios tardíos, por la progresión de la encefalopatía y las alteraciones cardiocirculatorias.
Otras determinaciones que nos pueden ayudar a clasificar las acidosis lácticas son: índice láctico/piruvato (L/P: redox citosólico) e índice 3OH-butirato/acetoacetato (3OHB/AA: redox mitocondrial)
- L/P normal (<20) o bajo (<10) sin cetosis: deficiencia de piruvato deshidrogenasa.
- L/P aumentado (>20) con 3OHB/AA aumentado: trastorno de la cadena respiratoria mitocondrial o mala extracción.
- L/P aumentado con 3OHB/AA normal o bajo: deficiencia de piruvato carboxilasa, deficiencia múltiple de carboxilasas (biotina) o deficiencia de alfa-cetoglutarato deshidrogenasa (ciclo de Krebs).
2.3.• Hiperamonemia:
Las hiperamonemias pueden ser:
2.3.1. Primarias: Trastornos del ciclo de la urea
2.3.2 Secundarias
- Acidurias orgánicas
- Trastornos del metabolismo de la biotina
- Trastornos del metabolismo del piruvato
- Deficiencia de la beta oxidación de ácidos grasos
2.3.3. Adquiridas
- Síndrome de Reye
- Tratamiento con valproato
- Ingesta de arginina insuficiente (malnutrición)
- Insuficiencia hepática
2.3.4. Hiperamoniemia transitoria del recién nacido: suelen presentar niveles de amonio menos del doble de lo normal.
2.4.• Hipoglucemia (<40 mg/dl): La clave ante una hipoglucemia es investigar si existe cetosis.
2.4.1.- Hipoglucemia no cetósica
- Hiperinsulinismo
- Defectos de la oxidación de los ácidos grasos:
- Fallo hepático
2.4.1.- Hipoglucemia cetósica
- Sepsis.
- Insuficiencia suprarrenal.
- Hipopituitarismo (puede asociarse con ictericia colestática).
Trastornos de la cadena respiratoria mitocondrial
- Defectos de la gluconeogénesis
- Glucogenosis
- Afectación hepática. Galactosemia.
BIBLIOGRAFIA
- Campistol J.(2007).Enfermedades neurometabólicas de presentación neonatal como causantes de trastornos del neurodesarrollo.Rev Neurol 44:19-25.
- Franco-Fernández ML, Bernardo AB.(2008). Trastornos metabólicos de presentación neonatal.En: Verdu A, Garcia A, Martínez B(Ed), Manual de Neurología Infantil,(pp190-199). Madrid:Publimed.
- García-Cazorla A.(2008).Enfermedades neurometabólicas: orientación para el neuropediatra. Rev Neurol 47:55-63.
- Gonzalez Gutierrez-Solana L, Rubio O. (2005). Manual de Urgencias en Neurología Infantil. En: Garcia-Peñas JJ, Gonzalez L, Ruiz-Falco ML (Ed). Manual de Urgencias en Neurología Infantil, (p455-480) España: Gráficas Enar.
- González Gutiérrez-Solana L, López Marín L, Cantarín Extremera V.(2010).Actitud ante la sospecha de un trastorno metabólico. Pediatr Integral. 14:396-404.
- Lyon G. (2006). The neurology of neonatal hereditary metabolic diseases. En: Lyon G, Kolodny EH, Pastores GM (Ed), Neurology of hereditary Metabolic diseases of children, (pp9-64). United States: McGraw-Hill.
- McCabe LL, McCabe ERB.(2008).Expanded newborn screening: implications for genomic medicine. Annu Rev Med.59:163-75.
- Nyahn WL. (2010). When to suspect metabolic disorder. En: Hoffmann GF, Zschocke J, Nyhan WL (Ed). Inherited Metabolic Diseases; a clinical approach, (pp 18-23). London New York: Springer.
- Nyahn WL. (2010). Metabolic emergencies. En: Hoffmann GF, Zschocke J, Nyhan WL (Ed). Inherited Metabolic Diseases; a clinical approach, (pp 25-34). London New York: Springer.
- Ruiz M, Santana C.(1998).Enfoque práctico para el diagnóstico de errores congénitos del metabolismo. Acta Pediatr Esp 56:39-52.
- Saudubray JM, Nassogne P, de Lonlay P, Touati G.(2002).Clinical approach to inherited metabolic disordes in neonates: an overview. Semin Neonatol.7:3-15.
- Sanjurjo P, Baldellou A, Aldámiz-Echevarría L.(2003).Enfermedades congénitas del metabolismo: bases diagnósticas para el pediatra. Madrid: Ergon.
- Shulze A, Linder M, Kohlmuller D.(2003).Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics.111:1399-406.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}